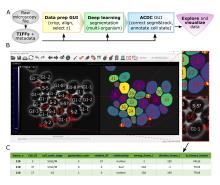
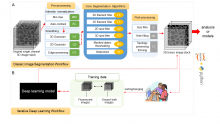
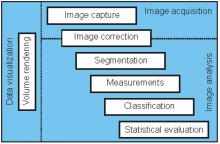
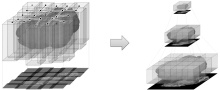
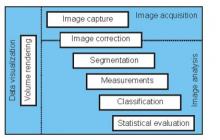
ilastik is a simple, user-friendly tool for interactive image classification, segmentation and analysis. It is built as a modular software framework, which currently has workflows for automated (supervised) pixel- and object-level classification, automated and semi-automated object tracking, semi-automated segmentation and object counting without detection. Most analysis operations are performed lazily, which enables targeted interactive processing of data subvolumes, followed by complete volume analysis in offline batch mode. Using it requires no experience in image processing.
ilastik (the image learning, analysis, and segmentation toolkit) provides non-experts with a menu of pre-built image analysis workflows. ilastik handles data of up to five dimensions (time, 3D space, and spectral dimension). Its workflows provide an interactive experience to give the user immediate feedback on the quality of the results yielded by her chosen parameters and/or labelings.
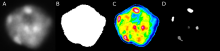
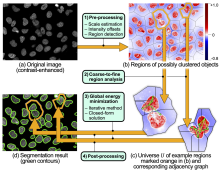
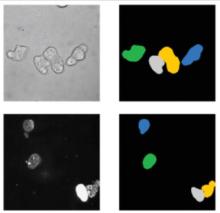
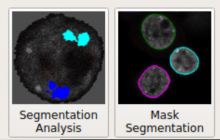
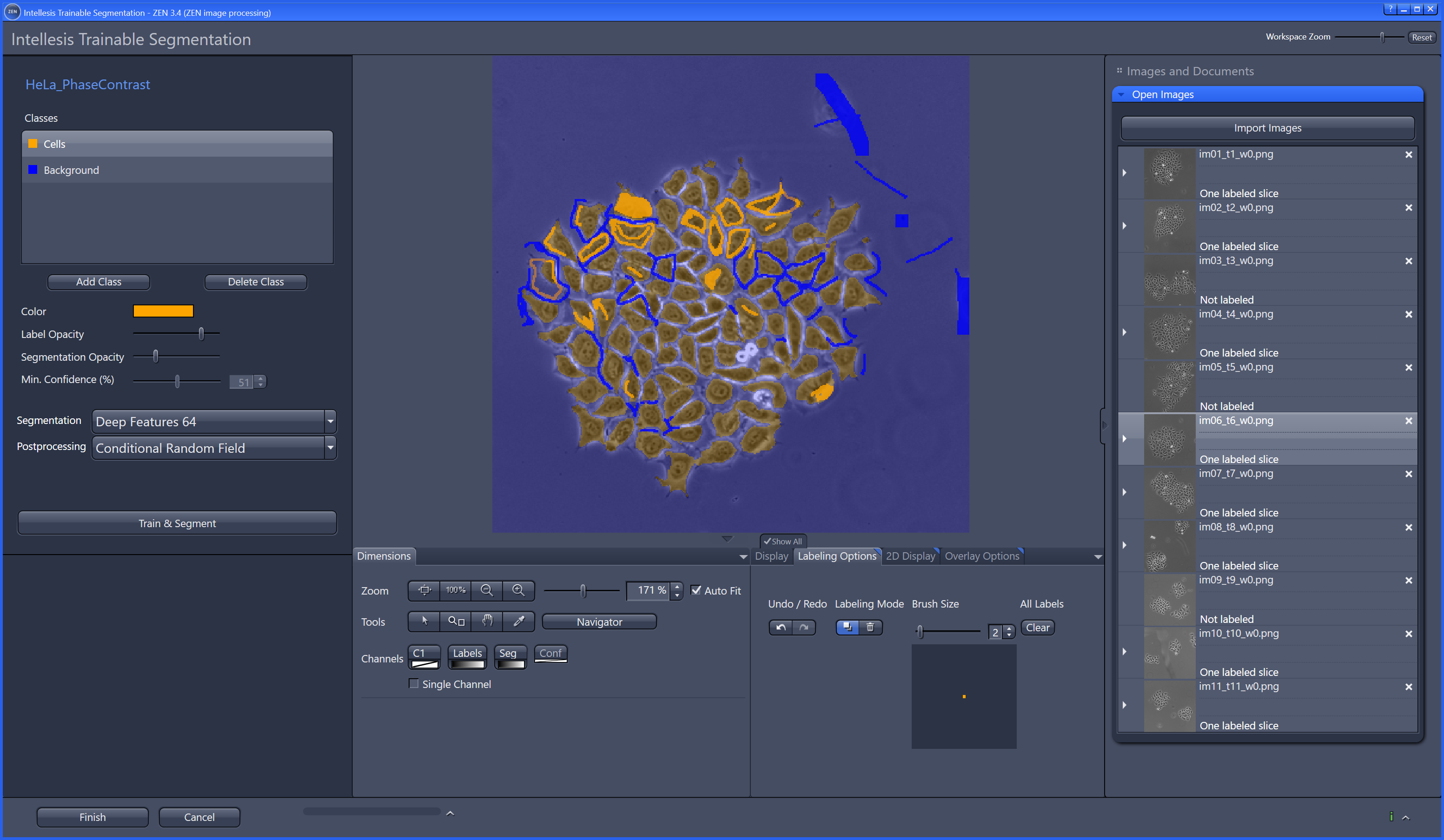
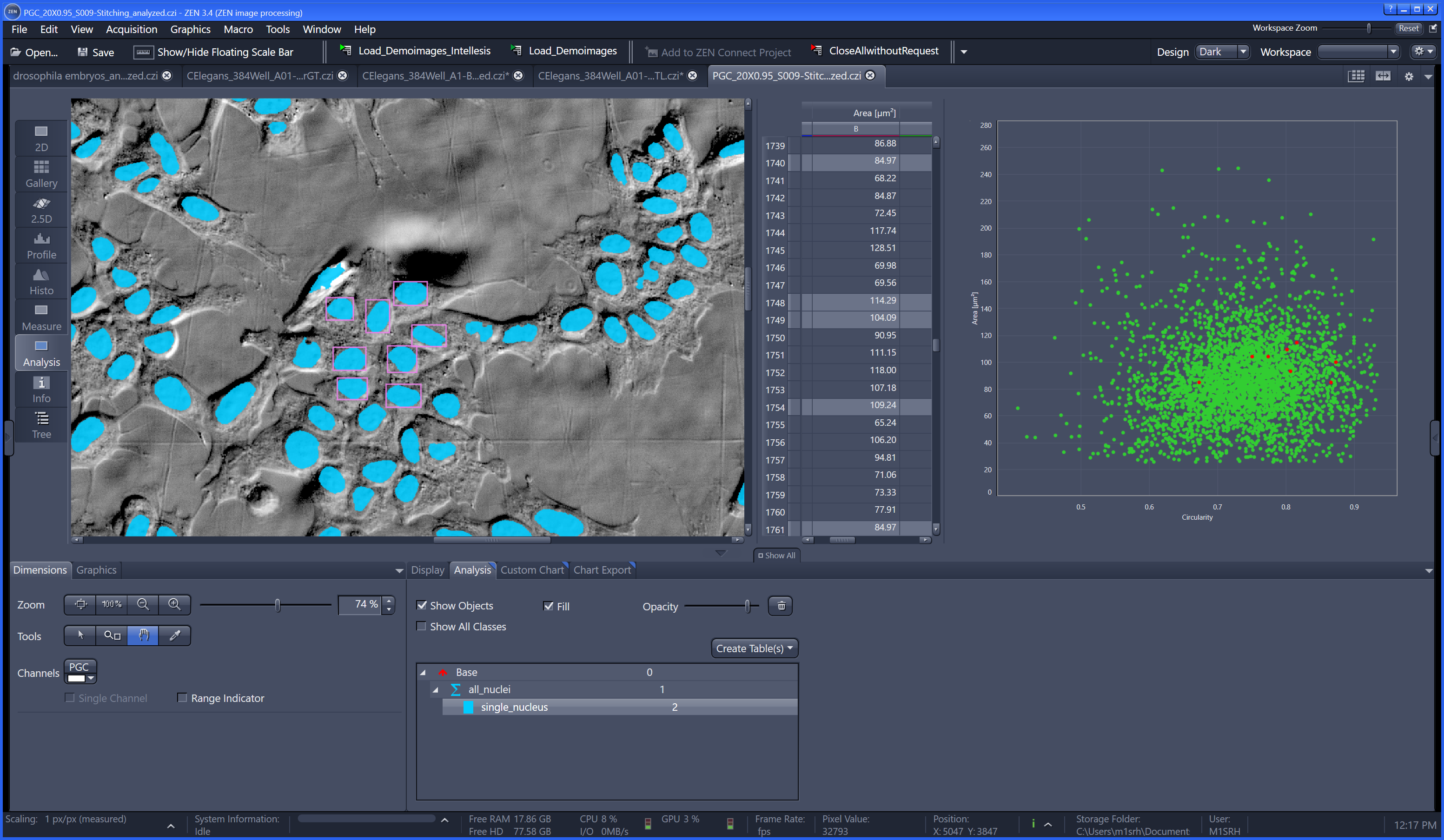
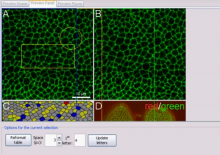
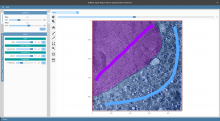
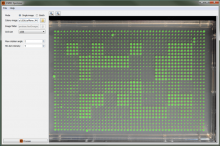
The most commonly used workflow is pixel classification, which requires very little parameter tuning and instead offers a machine learning technique for segmenting an image based on local image features computed for each pixel.
Other workflows include:
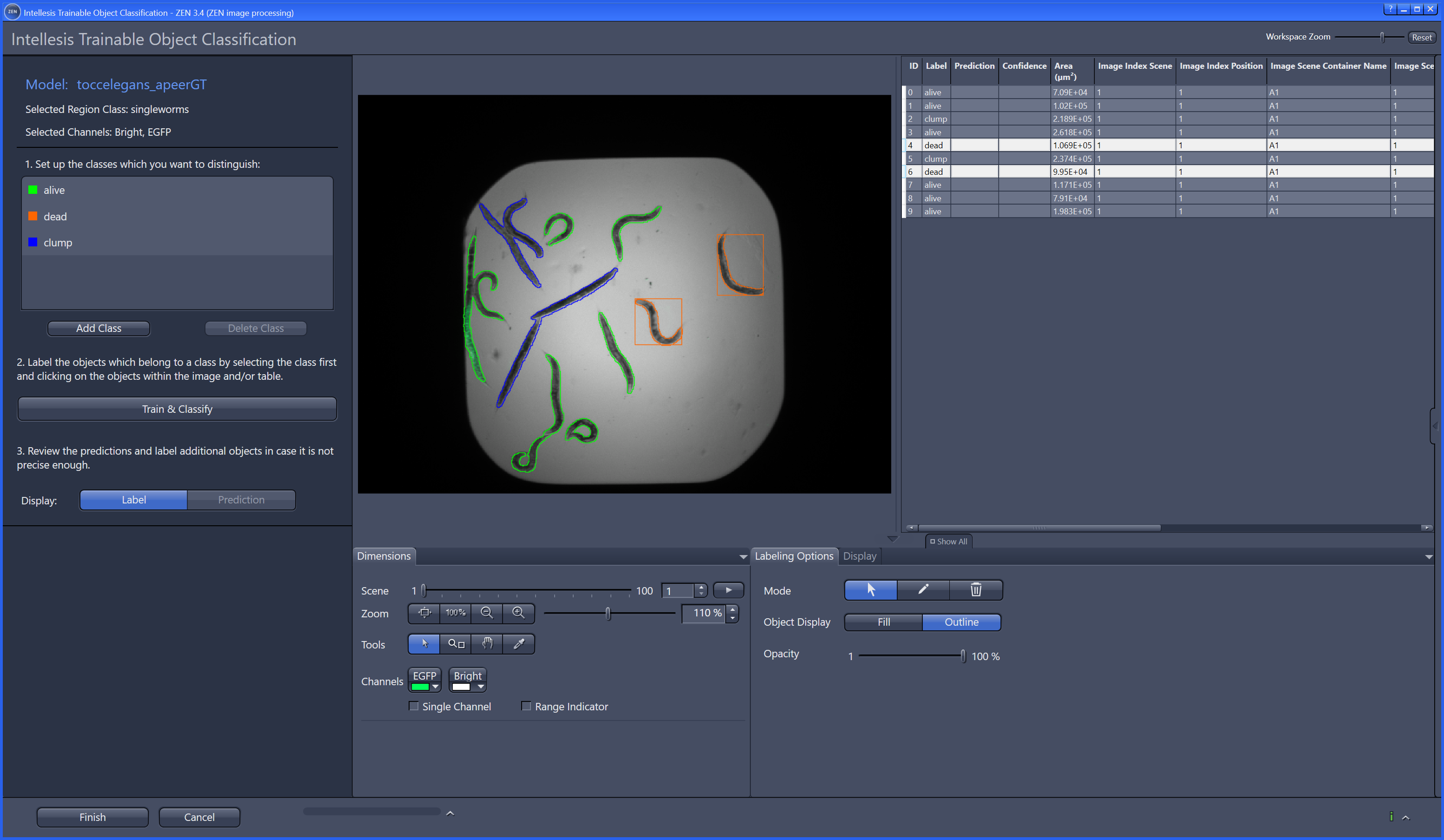
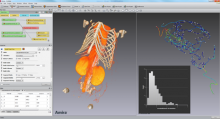
Object classification: Similar to pixel classification, but classifies previously segmented objects by object characteristics in a subsequent step
Autocontext: This workflow improves the pixel classification workflow by running it in multiple stages and showing each pixel the results of the previous stage.
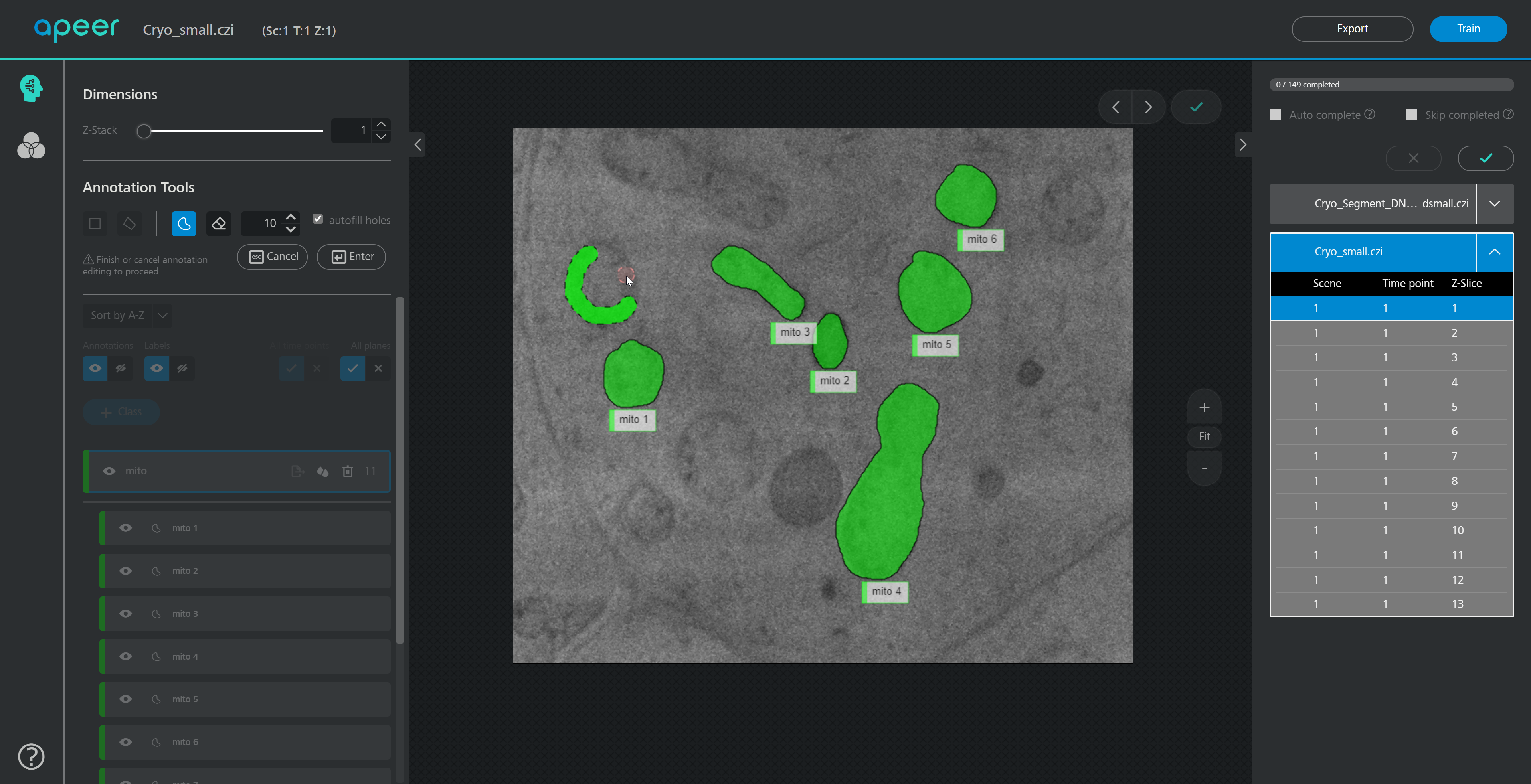
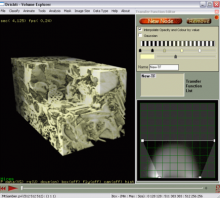
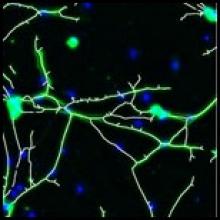
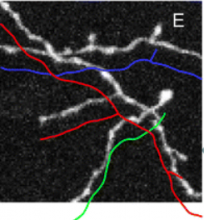
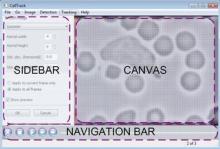
Carving: Semi-automated segmentation of 3D objects (e.g. neurons) based on user-provided seeds
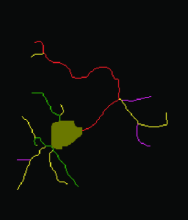
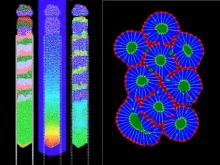
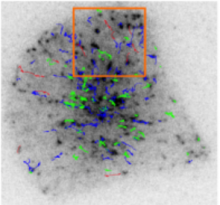
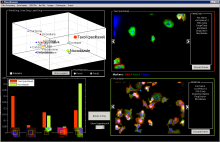
Manual Tracking: Semi-automated cell tracking of 2D+time or 3D+time images based on manual annotations
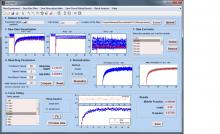
Automated tracking: Fully-automated cell tracking of 2D+time or 3D+time images with some parameter tuning
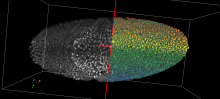
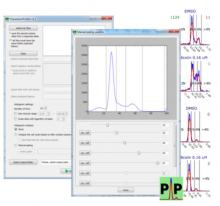
Density Counting: Learned cell population counting based on interactively provided user annotation
Strengths: interactive, simple interface (for non-experts), few parameters, larger-than-RAM data, multi-dimensional data (time, 3D space, channel), headless operation, batch mode, parallelized computation, open source
Weaknesses: Pre-built workflows (not reconfigurable), no plugin system, visualization sometimes buggy, must import 3D data to HDF5, tracking requires an external CPLEX installation
Supported Formats: hdf5, tiff, jpeg, png, bmp, pnm, gif, hdr, exr, sif