Description
Task
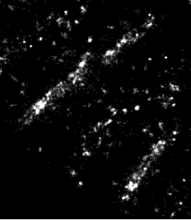
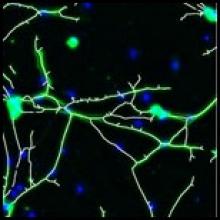
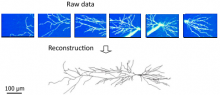
Quantify the length of microtubules (MT) and the MT average density per cell.
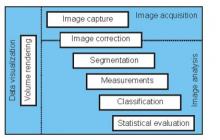
Workflow descriptions
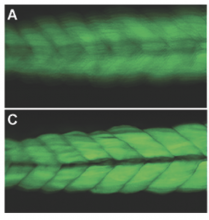
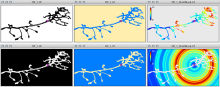
Simple two step workflow, allowing visual & manual correction of microtubule between the 2 steps. Batch measurement of microtubule lengths for multiple images is achieved by segmenting the MTs and then their skeletonizations. The number of pixels in the microtubule is proportional to their length, so the length can be estimated.
Script
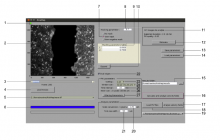
Workflow is written as an ImageJ macro (Fiji) with following steps:
1. The enhancement of tubular structure by computing eigenvalues of the hessian matrix on a Gaussian filtered version of the image ( sigma 1 pixel), as implemented in the tubeness plugin.
2. The tubules were then thresholded , and structures containing less than 3 pixels were discarded.
3. If needed, a visual check and correction of segmented microtubule is then performed.
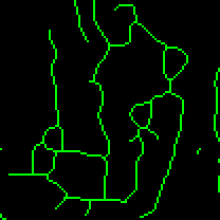
4. After correction, segmented MTs were then reduced to a 1-pixel thick line using the skeletonize plugin of Fiji. The length of the skeletonized microtubules was then directly proportional to their length.
5. Data were grouped by condition and converted back to micrometers units under Matlab for the statistical tests.
Pitfalls
Commented but not that general without editing some fields in the macros.
Sample Data
Sample data and workflow (see above URL) can be accessed by - login: biii - password Biii!
Misc
3D version also available here. Use of components Skeletonize and Tubeness Filter



































{kind=link}