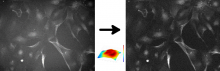
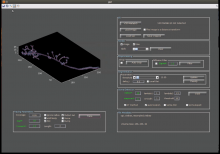
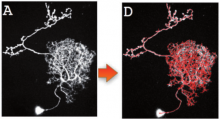
Description
Vaa3D is a handy, fast, and versatile 3D/4D/5D Image Visualization and Analysis System for Bioimages and Surface Objects. It also provides many unique functions that you may not find in other software. It is Open Source, and supports a very simple and powerful plugin interface and thus can be extended and enhanced easily.
Vaa3D is cross-platform (Mac, Linux, and Windows). This software suite is powerful for visualizing large- or massive-scale (giga-voxels and even tera-voxels) 3D image stacks and various surface data. Vaa3D is also a container of powerful modules for 3D image analysis (cell segmentation, neuron tracing, brain registration, annotation, quantitative measurement and statistics, etc) and data management. This makes Vaa3D suitable for various bioimage informatics applications, and a nice platform to develop new 3D image analysis algorithms for high-throughput processing. In short, Vaa3D streamlines the workflow of visualization-assisted analysis.
Vaa3D can render 5D (spatial-temporal) data directly in 3D volume-rendering mode; it supports convenient and interactive local and global 3D views at different scales... it comes with a number of plugins and toolboxes. Importantly, you can now write your own plugins to take advantage of the Vaa3D platform, possibly within minutes!