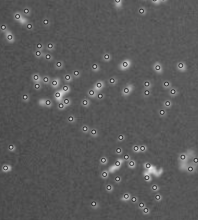
Description
This is a classical workflow for spot detection or blob like structures (vesicules, melanosomes,...)
Step 1 Laplacian of Gaussian to enhance spots . Paraeters= radius, about the average spot radius
Step 2 Detect minima (using Find Maxima with light background option to get minima). Parameter : Tolerance to Noise: to be tested, hard to predict. About the height of the enhanced feautures peaks